

|
|
|
 |
First described by the French neurosurgeon O. Crouzon in 1912, this condition includes an abnormal closure of growth plates (called sutures), which prevents normal growth of the skull and facial bones. Crouzon syndrome is currently thought to be the result of a change in a single gene, but currently no one knows what causes this to happen. In most cases, neither parent of a child born with Crouzon syndrome will have this condition, and the mother will have done everything right during her pregnancy. The process of bringing genes together from both parents is complicated and during this process changes in genes can normally occur. If a change happens to occur in one particular gene, the baby will be born with Crouzon syndrome. If neither parent has Crouzon syndrome, the chances of having a child with Crouzon syndrome has been estimated to be somewhere between 1:50,000 and 1:100,000 births. Crouzon syndrome can be inherited and is passed on in what geneticists call an autosomal dominant pattern. This means that if one parent has Crouzon syndrome, with each pregnancy there is a 50% chance of that the baby will be born with this condition.Today, it is possible for individuals who have Crouzon syndrome to elect to not pass on this trait on their children by undergoing in-vitro fertilization and selecting an embryo that does not have the Crouzon substitution for implantation. |
Types of Crouzon Syndrome:
There are two types of Crouzon syndrome: the classic form and Crouzon with acanthosis nigricans (a discoloration of roughened skin in the neck, armpits and groin). Crouzon syndrome occurs as the result of a mutation, or change, in a gene called FGFR2 located on the 10q chromosome. As it turns out, this same gene mutation can also result in a slightly different syndrome called Pfeiffer syndrome, which is distinguished by larger thumbs and big toes. It seems likely that other genes beside the FGFR2 gene may determine which syndrome occurs. Or, there could possibly be other factors outside of genes (called epigenetic factors) that influence which syndrome develops. Crouzon with acanthosis nigricans results from a mutation in a different gene, called FGFR3 on chromosome 4p. Meeting with a geneticist can help families better understand the genetics underlying these conditions.
Physical Traits:
The Skull
Children born with Crouzon syndrome have a slightly different appearance because the bones of the face and skull cannot grow in an entirely normal way. Parents may notice that their child’s eyes look bigger than other children, or that the forehead seems a little larger. Our skulls are made up of different bones and the junctions where these bones meet are called sutures (go to Craniosynostoses to see a picture of these sutures). In Crouzon syndrome at least two of these sutures (called the coronal sutures, running from ear to ear across the top of the head) are fused shut. Often as children get older, additional sutures will close abnormally. Early in life, sutures function as expansion joints to help the skull to grow. With the coronal sutures fused shut, as the brain grows it cannot “push” the forehead forward or the back of the head backwards. Instead, the brain pushes the skull to grow upward and wider.
The Brain
Some babies born with Crouzon syndrome can develop a condition called hydrocephalus. Deep inside the brain are open areas called ventricles that are filled with a water-like liquid called cerebral spinal fluid. These ventricles could be thought of as little lakes inside the head, which are connected to each other by small streams. Most children with Crouzon syndrome have ventricles that are larger than normal; however, this enlargment may not require treatment unless a pediatric neurosurgeon determines that the enlargement is associated with an increased pressure. Should this occur, a shunt, or tube, can be placed inside the ventricle and then tunneled under the skin down to the abdominal cavity in order to drain off the fluid. Typically, once a shunt is placed, the child will need this for the rest of his/her life. One possible alternative to placing a shunt is called an endoscopic third ventriculostomy (ETV). This operation opens up a connecting pathway between two different ventricles. When it works, it can avoid some of the potential complications associated with a shunt. The most obvious sign that a child might be developing hydrocephalus is when the head circumference measurements enlarge faster than would be predicted by the normal growth curves. For this reason, it is important to monitor head circumference measurements frequently in the first year of life. Most children with Crouzon syndrome do not develop hydrocephalus.
One more common condition that can occur with Crouzon syndrome is cerebellar tonsillar herniation, or a Chiari. There is a large hole in the floor of the skull called the foramen magnum, through which the spinal cord exits the skull. Sometimes the back part of the brain just above the foramen magnum, called the cerebellar tonsils, can get pushed down into this hole like a cork in a wine bottle. This can produce one type of sleep apnea, called central apnea. With this type of apnea the brain “forgets” to tell the body to take a breath. This condition can only be diagnosed by an overnight sleep study. Chiari malformations are usually not present at birth, but tend to slowly develop over time. We believe it is important that children with Crouzon syndrome undergo frequent screening for cerebellar tonsillar herniation and at our center in Dallas we have recently begun recommending occasional MRI scans even in adulthood. The development of a Chiari may be a sign for increased intracranial pressure, signaling the need to enlarge the skull to create more room for the brain (see Publication #19). Chiari malformations are difficult to assess on routine CT scans and are best evaluated with MRI scans. One study suggested that somewhere around 70% of children with Crouzon syndrome will develop a Chiari. However, not all Chiari’s require treatment, only those that cause symptoms. Some things we watch for include: difficulty swallowing, coordination issues, cough headaches, central sleep apnea, and the development of a syrinx (which looks like a widened river of fluid running down the center of the spinal cord). There are different surgical approaches for treating a Chiari malformation and parents are advised to seek second opinions at centers specializing in treating these rare conditions.
One constant concern for children born with Crouzon syndrome is the possibility for raised intracranial pressure. We know that children with Crouzon syndrome have larger brains. Although the skull can get bigger without sutures, the abnormal closure of skull sutures (craniosynostosis) does somewhat limit the ability for the brain to enlarge freely in all directions, which can lead to slightly higher pressures inside the skull, which in turn may reduce blood flow to the brain. Raised intracranial pressure is most often treated by surgically enlarging the skull (see Treatment below), an operation that usually has to be done somewhere between 2 and 4 times during a child’s lifetime, depending upon when the first operation is done and how well it is performed. Currently, no one is sure exactly what the ideal age is for the first skull enlargement operation. Studies have shown that most craniofacial surgeons perform the first skull enlargement around 6-months of age. Our research suggests that doing surgery at this early age is not only unnecessary, it is too soon because it impairs the future growth of the skull, often to the extent that additional operations will need to be added in the future. In Dallas, we believe it is safe to delay the first skull surgery well beyond 6-months of age to somewhere around 15-months of age, depending upon the degree to which the skull is affected. Not only are we convinced that this delay is safe, we are convinced that by waiting until a child is older before enlarging the skull, we can reduce the total number of lifetime operations. Sometimes parents are told that their child has raised pressure and must have surgery right away. It is very important for parents to find out exactly what has convinced their doctor that urgent early operations are necessary. Our experience suggests that it usually takes quite a while for raised intracranial pressure to have any adverse effects, and most often the biggest effect is on vision, not on mental development. However, it is important that growing children be closely monitored by experienced physicians to make sure that the intracranial pressure is not getting too high. This monitoring is done a number of ways: having a pediatric neuro-ophthalmologist evaluate the optic nerves (raised pressure can cause bulging of the nerve in the back of the eye), following head circumference measurements with growth, monitoring changes in the MRI scans, performing “visual evoked potentials” (a measurement of the speed that light travels from the eye nerve to the brain) or “optical coherence tomography” (a measurement of the thickness of the back of the eye), monitoring overall intellectual development, and looking for signs in older children such as headaches or vomiting. Lastly, it is even possible to put a tiny catheter under the skull to directly measure pressure.
Most children with Crouzon syndrome have normal developmental and intelligence. However, children who undergo multiple anesthetics and surgical procedures might be at greater risks for potential developmental delays. In addition, other factors such as chronic sleep apnea and abnomal brain development can also result in impaired development. I believe that the most important thing that those caring for children with Crouzon syndrome can to help children realize their full potential is to ensure that they do not have sleep apnea and to limit the number of lifetime operations. It is also critically important for parents to challenge their child to develop to his or her fullest.
The Eyes
Children with Crouzon syndrome usually have a downward slope of the eye fissure openings and appear to have bigger eyes. Actually, the eyeballs are normal in size, but what makes them appear bigger is that the bones surrounding the globes (called the orbits) cannot grow forward in a normal way. Sometimes the eyes can bulge so far forward (a condition called proptosis, or exophthalmos) that they becomes exposed, dry out, and can eventually become scarred. When the eyes are very prominent there is the risk that if a child cries too hard, the upper eyelid can become temporarily trapped behind the eyeball. Should this occur, one remedy is to put something like a wet Q-tip under the eyelid to pull it forward, back over the eyeball (see Initial Treatment below). Should this occur, it is possible to prevent this for happening again by doing a small operation called a tarsorrhaphy (sewing the outside part of the upper and lower eyelids together).
Most children with Crouzon syndrome have an imbalance of the muscles that move the eyes (called strabismus). Sometimes, one of the six muscles that move the eye may be completely absent. If the eyes do not line up in a straight forward gaze, children learn to stop seeing out of one eye (amblyopia) in order to prevent seeing with double vision. Should this occur, eye muscle balancing surgery may be needed.
The final long-term eye issue that can occur with Crouzn syndrome is called optic nerve atrophy (weakening, or wasting of the nerve that transmits sight), which can diminish a child’s ability to see. It is not known for sure what causes this optic nerve atrophy, but it is likely related to chronic elevations in intracranial pressure.
The Ears
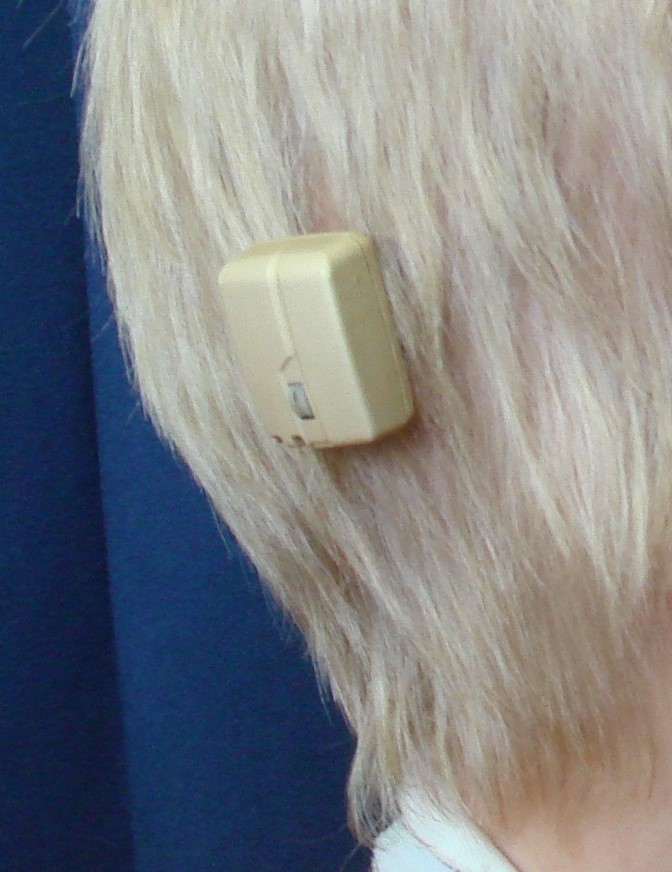 The external ears are usually normally shaped, but may be positioned slightly lower than average. The inner ear can be affected in a number of ways. As a result of poor facial growth, the inner ear does not drain well and children can develop fluid in their ears (serous otitis) as well as more frequent ear infections. Most infants require placement of tubes in the ears to equalize pressure and to improve hearing. In addition, the bones of the inner ear may be partially fused together, contributing to a conductive hearing loss. Rarely, children might be candidates for BAHA’s (bony anchored hearing aids, which are implanted directly into the skull). We recommend delaying implantable BAHA placement until children are in their teens because the presence of BAHA’s can complicate certain types of skull remodelling that is often required in adolescence. Some adults with Crouzon syndrome can have partial permanent hearing loss, so careful attention to the ears is important in infancy and childhood.
The external ears are usually normally shaped, but may be positioned slightly lower than average. The inner ear can be affected in a number of ways. As a result of poor facial growth, the inner ear does not drain well and children can develop fluid in their ears (serous otitis) as well as more frequent ear infections. Most infants require placement of tubes in the ears to equalize pressure and to improve hearing. In addition, the bones of the inner ear may be partially fused together, contributing to a conductive hearing loss. Rarely, children might be candidates for BAHA’s (bony anchored hearing aids, which are implanted directly into the skull). We recommend delaying implantable BAHA placement until children are in their teens because the presence of BAHA’s can complicate certain types of skull remodelling that is often required in adolescence. Some adults with Crouzon syndrome can have partial permanent hearing loss, so careful attention to the ears is important in infancy and childhood.
A BAHA hearing aid.
The Midface
The midface is the part of the face that extends from underneath the eyes down to the teeth of the upper jaw. This part of the face is always smaller than normal (also called hypoplastic, meaning lack of growth), with the deepest part of the face typically centered at the top of a foreshortened nose. Because the midface does not grow forward in a normal way (actually, it is smaller in 3-dimensions), most children need to have their midface brought forward with an operation (see Treatment below). Based on measurements taken from our center, it appears that the face only grows about 1/3 normal speed, although this varies from child to child. There is additional evidence to suggest that all forward growth stops about age 9 (see Publication #25) or immediately following any operation performed to advance the midface.
The Mouth, Palate, and Airway
In addition to the midface not growing forward in a normal way, the palate, or roof of the mouth, has a very high arch. This high arch pushes up the floor of the nose. Not only does reduce the size of the nasal airway, parents may notice that their toddler’s noses seem to “run” all the time. This problem usually gets better as children grow into their teens.
The airway in Crouzon syndrome is usually smaller than normal and parents may notice that their children are noisy breathers, especially at night. With the palate arching upwards, the nasal passages are squished smaller. In addition, the roof of the mouth may be lower in the back, almost touching the tongue, making the oral airway smaller. Sometimes the tongue will be slightly floppier in babies, which allows it to fall back and block the airway. For this reason, parents may notice that their child breathes better when positioned on their side, or stomach.
The windpipe, or trachea, is kept open by C-shaped rings of cartilage, similar to the rings of metal in the tubing that exits the back of a clothes dryer. This ring is C-shaped so that when we cough or breath deeply, it can expand. In Crouzon syndrome sometimes these rings may not open normally, or can be O-shaped (they start out as a “C,” and then close up to form an “O”). Rarely, the trachea maybe slightly narrowed at one spot; further contributing to a breathing difficulty. For this reason, all patients treated by our team in Dallas that have signs for airway problems will undergo an examination under anesthesia (this procedure is called a laryngoscopy, upper endoscopy or bronchoscopy). This is frequently performed at the same time as some other scheduled procedure in order to reduce the total number of anesthetics and operations. Children may also need to be seen by a pediatric pulmonologist (lung specialist) if there is any suggestion for asthma. Most importantly, infants and children must be routinely tested for apnea with sleep studies, in many instances on at least a yearly basis (see Treatment).
The Abdomen
Based on our experience in Dallas, we believe that children with Crouzon syndrome might be a a slightly higher risk for having a condition called a malrotation (the intestines are twisted in an abnormal way). When this occurs, the child is at risk for developing an intestinal obstruction, or blockage. In addition, the appendix is abnormally displaced to the left upper stomach (instead of the right lower stomach). This condition is diagnosed with a test called an upper G.I. and when present, needs to be surgically treated by a pediatric surgeon.
Treatment
The care of a child born with an unusual condition such as Crouzon syndrome is best done a major craniofacial center by an experienced comprehensive team. The Craniofacial Center in Dallas has a special interest in Crouzon syndrome and we believe it is the busiest centers in the U.S. for treating this condition. Our specialists are willing to offer advice to parents and/or to other treating physicians. The following is a brief overview of our recommended treatment protocol in Dallas; however, specific recommendations for each child can only be made after a comprehensive examination and after reviewing appropriate test results (see Choosing a Doctor).
Initial Treatment
The most critical issues in initially caring for children born with Crouzon syndrome are:
1. Making sure that there are no problems with breathing, 2. Protecting the eyes from damage, and 3. Ensuring adequate nutritional intake. For those more severely affected infants, a tracheostomy might be recommended right after birth. Otherwise, consideration might be given for a child to go home on an oxygen saturation monitor (which has an alarm that goes off if a child is not getting enough oxygen at night). Although many centers request a CT scan as a first test, these are typically not performed at our center because we do not believe that these scans have any bearing on how treatment should be begun. Instead, the first test our center will recommend is a sleep study. This test is performed to determine if there is sleep apnea and to ensure the child is getting enough oxygen at night to maximize brain development. Some children may also need to be started on medication to help reduce gastro-esophageal reflux (GER). Unrecognized, severe reflux can even cause infants to briefly stop breathing and turn blue.
In terms of eye protection, early “permanent” tarsorrhaphies might be needed. This small operation entails sewing the upper and lower eyelid together near the outside corners of the eye. This is actually not permanent and is a completely reversible operation (if done in the proper way). For children with large bulging eyes, I recommend that parents keep a tube of Lacrilube or Genteal ointment at home to moisturize the eyes until a tarsorrhaphy can be performed. Should your child’s eyelid get trapped behind the eye, if you are unable to quickly pull the upper eyelid back over the globe (with a wet Q-tip under the eyelid) then put lots of ointment over the entire eye (to prevent drying and scarring) and go directly to the emergency room.
Skull Surgery
The timing for the first skull operation varies significantly depending upon how the skull is growing. A general rule of thumb is to delay the skull surgery for as long as possible. Children treated in Dallas might be delayed until 15-months of age, or even older, depending on multiple factors. The reasons for this delay are complicated but one concern is that our operations will impair skull growth, which means that if surgery is done too soon can mean more operations will be necessary later. As babies grow and get bigger, their total blood volumes go up; we believe that this lessens the likelihood that a blood transfusion will be necessary and also that operations may be less risky as babies grow older. We routinely recommend that children receive a drug called erythropoietin before surgery to raise blood levels. We have published a study that showed that those getting this drug before surgery had a much lower risk of needing blood transfusions (see Publication #20). We also use a “cell-saver” to recycle much of the blood that is lost at surgery, so that it can be given back to the child during the operation (Publication #23). Using this combination of techniques, most children (now well over 90%) do not require any blood transfusions. We believe that reducing blood transfusions also reduces the risks of other serious complications.
At our center operations on the skull are always performed by a pediatric neurosurgeon and craniofacial surgeon working together over the course of the entire procedure. We believe that having two experienced surgeons present at all times improves the speed and safety of the operation. In addition, only pediatric anesthesiologists with extensive craniofacial surgical experience are used. One of our pediatric ENT’s will often evaluate the child in the operating room and place ear tubes at the same time as skull surgery, in order to save having to do an additional operation. Typically, skull surgery takes two hours, but the children are in the operating room for 4 to 4 ½ hours total. Whether or not the front half of the skull is brought forward (anterior CVR or FOA), or the back half moved further backwards (posterior CVR), depends upon the findings on a preoperative MRI scan. We do not shave any hair, although many centers still prefer to do so. Many years ago, I changed the typical straight-line incision to a wavy zigzag incision because I learned that when children get their hair wet, it will part right on a straight-line scar making it obvious (see Publication #10). Designing the incision in a wavy pattern helps to better hide the scar, especially when the hair is wet.
The goal of the first skull surgery is to enlarge the skull to give the brain more room to grow. Once a suture is fused shut, it cannot be “released” in such a way as to permit better growth. This is because it is impossible to surgically recreate a functioning suture; therefore, it is important that the surgery is able to accomplish a significant enlargement. Surgeons use various techniques to hold the skull bones in place after moving them into position. We have previously reported our findings that because of the way that the skull grows, when other surgeons use metal plates and screws to put the skull back together these plates may eventually end up on the inside of the skull with the screws poking into the brain (Publication #12). While I am unaware of any cases in which this has caused a problem, I have nevertheless chosen to only use dissolving stitches to put the skull back together (Publication #21). With this technique, nothing artificial is left behind as the child grows. Dissolving plates and screws can also end up inside the skull of growing infants and can weaken the skull bones, making subsequent operations more technically challenging. Rarely, in a tiny percentage of cases, the dissolving plates and screws will melt into a liquid that may drain out through a small hole in the overlying skin. So, while it is technically a little more difficult to just use dissolving sutures to rebuild a skull, we believe this technique provides the best results with the fewest complications.
Many years ago, with the hope of being able to enlarge the skull more than with traditional skull surgery, I evaluated using distraction devices with metal rods poking out of the head that were used to slowly expand the skull. I found that not only was skull distraction difficult for both the child and parents; it also required two operations instead of one (a second operation is needed to remove the distraction device months after the first operation). I also found that distraction deformed the skull, had a higher complication rate, and didn’t seem to provide any better result than doing just a single operation. Therefore, I do not currently recommend this technique for my patients. I also believe that helmets or headbands should never be used on a child with craniosynostosis. These restrict skull growth, which should never be done in Crouzon syndrome.
At the end of the operation in Dallas, the scalp is closed with dissolving stitches. We never use metal staples, nor do we use drainage tubes, because both hurt when they are removed. We also do not put any bandages on the child. Instead, we just wash the hair and comb it out before leaving the operating room.
In my opinion, the most critically important step at the end of an operation to enlarge the front half of the skull, particularly for children with prominent eyes, is for the surgeon is to place a dissolving suture that keeps the eyelids partly closed, but not so much that the child cannot see. We have unfortunately seen a number of children operated at other centers who did not get these eyelid stitches and as a result of swelling after surgery, ended up completely losing vision in one or both eyes.
Children typically will spend one night in the pediatric intensive care unit before being transferred to the floor the following day. We encourage parents to hold their child in their laps, versus in a crib, after surgery. Today, no narcotics are given to children after surgery; instead, we keep children comfortable using intravenous acetaminophen and ibuprofen (Publication #58), which we have found also reduces nausea and vomiting after surgery. At our center most all children spend only two nights total in the hospital prior to discharge. The risks of the surgery are very small at experienced centers. Many studies have shown that surgeons with the most experience tend to have the fewest complications (Publication #37). We have published a two-center study (Publication #15) that showed no infections occurred in infants undergoing operations for the first time (however, it is still possible for this to occur).
Treating Sleep Apnea
A small degree of sleep apnea can be considered normal, although the amount varies with age. There are also two kinds of sleep apnea: central and obstructive. Central apnea occurs when the brain skips a breath and is most commonly caused by cerebellar tonsillar herniation, or a Chiari malformation. But, in rare instances central apnea can be a sign for raised intracranial pressure. One treatment for central apnea is to enlarge the back of the skull, while simultaneously enlarging the bone around the upper spine to decompress the Chiari (Publication #48). The other type of sleep apnea, or obstructive apnea, is the most common type of apnea in Crouzon syndrome and is the result of a narrowed or blocked airway.
The first treatment for obstructive sleep apnea might simply be medication. There are a number of drugs that can open up a partially blocked airway to facilitate breathing. The next step might be to consider removing the tonsils (but only if they are significantly enlarged). Usually, taking out the tonsils only provides a modest improvement. The next step is a CPAP or BiPAP mask, which is worn at night and can be very effective when tolerated. If none of the above treatments are successful and the child is still young then the safest option, and best next step, might be a temporary tracheostomy. Understandably, no parent wants their child to have a tracheostomy; however, for some children, going without a tracheostomy can impair mental developmental and can even be life-threatening when the child gets sick with a severe cold or flu. When apnea is present in older children (in Dallas we recommend a child be at least 6 years old, but ideally 8 or 9), advancing the midface will provide the greatest improvement in breathing.
Surgery of the Midface
There are many different types of operations used to bring the midface forward in Crouzon syndrome, the Le Fort I, the Le Fort III, the monobloc and the bipartition. The LeFort I brings the lower half of midface forward from the floor of the nose down to the upper teeth. The Le Fort III brings the entire midface forward from the top of the nose and the cheekbones, down to the upper teeth. The monobloc brings the forehead and the midface together at the same operation. There is also a variation of the monobloc, called a bipartition, which splits the face down the middle. The Le Fort I is not recommended until children are in their teenage years, leaving the LeFort III and the monobloc the two most commonly performed operations. Among the advantages of the monobloc is that the forehead and midface are brought forward at the same time, saving one operation. However, the monobloc is not able to vertically lengthen the midface, it is not as good at treating obstructive apnea, and it does carry a higher risk for serious infections (also CSF leaks from the brain). For these and other reasons most surgeons, including myself, choose to not perform the monobloc (Publications #8, #66). Your surgeon will discuss what is he or she thinks is best for your child.
The two indications for advancing a midface are to treat obstructive sleep apnea that cannot be treated by any smaller means, or when appearance becomes an issue for the child (usually sometime after age 7). This operation is one of the bigger operations a child will undergo and probably has the highest complication rate of any of the other operations needed to treat Crouzon syndrome. For this reason, whenever possible families should consider taking their child to the most experienced surgeon they can find.
The Le Fort III procedure is performed by reopening the same incision used for the skull surgery. With the skin pulled down, a cut is made along the top of the nasal bones, across the floor of both orbits (under the eyes) and down the sides of both cheekbones. At our center, no scars are put on the child’s face. Since 1998, I have been using a halo-distraction technique, developed in Dallas, to advance the midface. After the bones behind the face have been cut, the incision on the top of the head is closed and a device called the RED (Rigid External Distraction) is applied on the outside to help bring the midface forward (Publications #18, #25, #55). This device is usually attached to a U-shaped splint on the upper teeth. While some surgeons will place wires through the skin of the face to attach to the underlying bone, I have found that is it possible to do this without leaving any permanent facial scars. The parents, or the child, then turn two screws on the halo every day in order to slowly (and painlessly!) bring the midface forward. Children are allowed to eat soft foods, may go to school and can even go swimming while wearing the RED. Approximately eight weeks later, the device is removed with a 20 minute anesthetic. Thus far, we have treated well over 175 children with the RED device and believe that we have learned many ways to make this process safer and easier. Today, the average length of hospital stay for a Le Fort III in Dallas is 3 to 4 nights.
 |
 |
 |
|---|
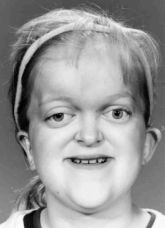
One example of a good result following a RED halo-distraction LeFort III on a young girl with a condition almost identicle to Crouzon syndrome, Pfeiffer syndrome, seen before surgery (left), 3 years postoperatively (middle) and over 6 years postoperatively (right).
The Big Picture
Most children born with Crouzon syndrome spend too much time in the hospital. We now know that it is possible to treat this condition with fewer operations, fewer nights spent in the hospital, and more time growing up as a normal child. However, if the total number of operations is to be reduced, it is critically important that the correct operation be done in the best way and at the right time. Treatment recommendations will vary from center to center and are constantly changing over time. It is important that parents make sure that when they meet with their doctors that they get all their questions completely answered. The Choosing a Doctor section can help families put together questions to ask their local doctors. Parents should not feel rushed into treatment. Take the time to learn as much as you can, get more than one opinion, and search until you find that physician that you feel has the experience to safely get the best results and the caring to coordinate all the various treatments that your child will need. In general, the busier the center, the lower the complication rates.
.
Jeffrey A. Fearon, MD, FACS, FAAP
Director, The Craniofacial Center, Dallas Texas
972-566-6464
Cranio700@gmail.com