
Apert Syndrome
Apert Syndrome Overview
Apert syndrome was first described in 1906 by a French pediatrician, Eugène Apert. It results from a change, or mutation, in a single gene located on chromosome 10q. As it turns out, there are two different mutations can cause Apert syndrome: Ser252Trp (S252W) and Pro253Arg (P253R). These mutations may lead to slightly different physical traits, but it is not always possible to identify the mutation type through examination alone.
The cause of Apert syndrome is unknown. In nearly all cases, both parents have normal genes, and a mother does “everything right.” Bringing together two parents’ genes to make a new baby is complex, and if a mutation occurs in just one spot, Apert syndrome can result. Research suggests that the mutation often comes from the father’s side, even though he does not have this condition.
The chance of two unaffected parents having a child with Apert syndrome is somewhere between 1 in 60,000 and 1 in 120,000. It is not possible to have Apert syndrome without knowing it, as the condition is always physically apparent.
When an adult with Apert syndrome has children, the condition follows an autosomal dominant inheritance pattern, which means that there is a 50% chance of passing it on with each birth. If both parents have Apert syndrome, there is only a 25% chance of not passing on the condition. Today, individuals can prevent transmission through in-vitro fertilization and genetic testing, selecting embryos without the Apert mutation for implantation.
Physical traits
Apert syndrome affects multiple areas of the body, and symptoms vary among individuals. Certain traits are more likely depending on the mutation type:
- S252W mutation: Higher likelihood of cleft palate
- P253R mutation: Increased chance of “rosebud” hand (thumb fused with fingers)
Other genetic factors may also influence the presence of specific traits.
Skin
Parents may notice that their child sweats more than other children. This condition is called hyperhidrosis and is caused by overactive sweat and oil glands. During adolescence, severe acne may develop. This can be effectively treated with medications such as Accutane®, which can help to prevent permanent scarring.
The Skull
The skull is made up of separate bones and the spaces between these bones are called sutures (if you want to see a picture of these sutures go to the Craniosynostoses section). In Apert syndrome, starting with the coronal sutures (running across the top of the head from ear to ear), most all of the sutures of the skull will eventually fuse shut. Once closed the skull cannot grow normally from front-to-back, causing the brain to grow upward and making the head appear taller.
Sometimes, the metopic suture, which runs vertically down the middle of the forehead, will split open like a seam tearing apart on a tight pair of pants. This will leave a large V-shaped soft area that can be felt, which later eventually closes up with bone.
With Apert syndrome, three different skull head shapes can occur:
- Type I: Wide-open soft spot, skull not very tall
- Type II: Taller skull, may have a ridge on the forehead
- Type III: Very tall skull with recessed forehead
We believe that the type of skull shape should determine the timing for skull surgery, and this is further discussed in the Treatment section.
The Brain
The brain contains fluid-filled ventricles, which are kind of like small lakes inside the head that are filled with what is called cerebral spinal fluid (or CSF). Most children with Apert syndrome have bigger ventricles, but this usually does not require any treatment. However, should they fill up with too much fluid they can begin to squish the brain from the inside. This condition is called hydrocephalus, and when this happens a neurosurgeon must then decide if it is better to place a shunt (small tube to drain off the extra fluid), or in selected cases, a third ventriculostomy (ETV). Our research has found most children with Apert syndrome (over 80%) never develop true hydrocephalus (Publication #47). However, we have noted that a higher percentage of children coming to our Center after having first been treated somewhere else have shunts, compared with those who began treatment at our Center. This suggests that centers having limited experience treating this condition might be mistakenly believing that these larger ventricles, which are normal in Apert syndrome, must be treated. We have learned that if a baby’s head circumference continues to follow their established growth curve, they usually do not have hydrocephalus.
Another condition that may affect the brain is called cerebellar tonsillar herniation, or a Chiari malformation. The floor of the skull has a hole called the foramen magnum, through which the spinal cord exits. Sometimes, part of the brain (the cerebellar tonsils) can get pushed down into this hole like a cork in a wine bottle. When this happens this Chiari can reduce the flow of cerebral spinal fluid around the brain stem and can cause a particular type of sleep apnea called central sleep apnea. With central apnea, a child will every now and then forget to breathe while asleep, lowering oxygen levels to the brain. There is no way to tell if this is happening by watching your child sleep, it is necessary to test for this by doing an overnight sleep study. A Chiari can also lead to a condition in the spinal cord called a syrinx, or syringomyelia, where the center of the spinal cord gets enlarged with fluid causing a small bulge in the cord. Although Chiari malformations were once believed to be very rare in Apert syndrome, we have found that this condition is actually much more common than originally thought, occurring in almost 30% of all our patients in Dallas (Publication #47). We believe that it is important to not just check for a Chiari early in life, but to continue to monitor for the development of a Chiari because these can develop over time. In addition, we have learned that the development of a Chiari may mean that there is an increase in intracranial pressure, signaling the need to enlarge the skull to create more room (Publication #19). Unfortunately, there are no outward signs that might be obvious to a parent if their child developed a Chiari; the only way is by specialized imaging. At the current time most craniofacial surgeons routinely order CT scans on their patients with Apert syndrome. However in Dallas, we have pretty much stopped getting CT scans for a number of reasons. To begin with, it is very hard to tell if a child might have a Chiari from looking at a routine CT scan; MRI scans are much better for evaluating the brain, especially in the region of the brainstem. We have also learned that by doing a careful physical examination it is possible to tell which sutures of the skull are fused without any x-rays (Publication #28). Finally, there have been a number of good studies that suggest that the radiation associated with CT scans might slightly increase the risk for brain and blood cancers over a child’s lifetime (http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(12)60815-0/fulltext) and there is no radiation with an MRI. In addition to looking at the ventricles and seeing if a child has a Chiari, we also look for other potential changes in different parts of the brain. Children with Apert syndrome may have a poorly formed region called the septum pellucidum, or another area that may not form normally called the corpus callosum. Interestingly, a review of our Apert patients suggests that the absence of these structures does not seem to correlate with a child’s mental development (Publication #47).
One constant concern for children born with Apert syndrome is the possibility for raised intracranial pressure. We know that children with Apert syndrome have larger brains. Although the skull can get bigger without sutures, the abnormal closure of skull sutures (craniosynostosis) does somewhat limit the ability for the brain to enlarge freely in all directions, leading to slightly higher pressures inside the skull, which in turn may reduce blood flow to the brain. Raised intracranial pressure is most often treated by surgically enlarging the skull (see Treatment below), an operation that usually has to be done somewhere between 2 and 4 times during a child’s lifetime, depending upon how early the first operation is performed and how well it is done.
Currently, no one is exactly sure what the ideal age is for the first skull‑enlargement operation. Studies suggest that most craniofacial surgeons perform the first skull enlargement around 6 months of age. Our research suggests that doing surgery at this early age is too soon (Publication #98). This is because surgery hurts the future growth of the head, meaning more operations will be necessary later. In Dallas, we believe it is safe to delay the first skull surgery up to around 15 months of age, or even longer depending upon the skull type (Type II and III skulls need to be expanded before Type I, closer to a year of age). Not only do we feel this delay is safe, we are convinced that waiting until a child is older before enlarging the skull can reduce the total number of lifetime operations.
Sometimes parents are told that their child has raised pressure and must undergo surgery right away. It is very important that parents first determine exactly what has convinced their doctor that an urgent operation is really necessary. Surgeons with less experience may worry about elevated intracranial pressure well before they need to be concerned, and performing surgery before it is necessary can cause more harm than good. How can doctors find out for sure if there is intracranial pressure? This can be done in several ways:
- An eye doctor (ideally a neuro‑ophthalmologist) examines the optic nerves. When pressure is elevated inside the head, the nerves in the back of the eye will appear to be bulging. When found (this is called papilledema) it indicates the need for surgery, and surgery should be probably be done within the next month or two. However, eye exams are not completely reliable; sometimes the eye exam can look normal even when there is elevated pressure.
- Follow head circumference measurements. A falling head circumference can raise the concern that intracranial pressures might be developing.
- Perform Visual Evoked Potentials (VEP’s). This test measures the speed of signals going from the eye to the brain. However, this test can be unreliable and therefore is infrequently used.
- Perform Optical Coherence Tomography. This test measures the thickness of the back of the eyeball. Few centers use this test, but it is considered to be more reliable than VEP’s.
- Monitoring changes in MRI scans. More experienced centers use these scans to determine if there is elevated pressure. The MRI must be done in a special way to permit measuring the widths of the optic nerve sheaths (Publication #91). At our Center in Dallas, we believe this is the best test currently available.
- Direct intracranial pressure testing. This involves a short operation to place a tiny catheter inside the skull to directly measure pressures for one or two days. It is considered the most accurate way to determine if there is intracranial pressure. Some centers might suggest a quick measurement of pressure (“spot check”) via a spinal tap, but we believe this test has limited indications.
Most children with Apert syndrome have some level of developmental delay. While frequent hospital stays and surgeries can play a part, they are not the only reasons. Other factors like sleep apnea, and differences in how the brain develops, may also contribute. An earlier review of 80 children with Apert syndrome coming to our center showed an average I.Q. score of 78; but scores varied a lot from child to child. Today, children with Apert syndrome are likely have higher average scores, thanks to improvements in care. A later study of over 130 patients treated in Dallas suggested that development was not influenced by how old children were when they had their first skull surgery (Publication #47). It also showed that children treated at our center from birth had higher developmental levels than those who began treatment elsewhere, although many factors may have contributed to this difference. Some children with Apert syndrome can have I.Q. levels in the low-to-normal range and go on to attend college. But most importantly, an IQ test measures only one type of intelligence. Children can be talented in many other ways that these tests don’t capture, such as musical ability, creativity, social skills, or artistic strengths. For children with Apert syndrome, one of the most important things caregivers can do is make sure they are not struggling with sleep apnea and to avoid unnecessary surgeries over their lifetime. We encourage parents to support and challenge their children so they can grow, learn, and reach their fullest potential.
The Eyes and Mid Face
Children with Apert syndrome often have eyes that appear larger and tilt slightly downward. Actually, the eyeballs themselves are normal in size. They just look bigger because the bones around the eyes (the orbits) don’t grow forward as they typically would. In some children, the eyes can bulge forward more than usual, a condition called proptosis or exophthalmos. When this happens, the surface of the eye can dry out, which may lead to irritation or scarring that can affect vision. In more severe cases, when a child cries, the eyelid can momentarily slip behind the eye. If this happens, gently placing something soft and moist, like a wet cotton swab, under the eyelid can help pop it back into place. If this problem happens repeatedly, a small procedure called a tarsorrhaphy can help. This involves partially stitching the outer corners of the eyelids together to protect the eye.
Some children with Apert syndrome may also develop optic nerve atrophy, which means the nerve that carries visual information to the brain becomes weaker. Our research suggests this happening in only 8% of children with Apert syndrome, and it seems that the presence of a Chiari might make this more likely (Publication #91). When this occurs it can affect a child’s vision. The exact cause isn’t fully understood, but it may be related to long‑term increases in pressure inside the skull.
The most common eye issue in children with Apert syndrome is strabismus, which means the eye muscles don’t work together the way they should. In some children, one of the six muscles that normally help move the eye, called the superior oblique muscle, may not be there at all. Because of this imbalance, many children need eye‑muscle surgery to help the eyes work together and to prevent amblyopia, a type of vision loss that can occur when the brain starts to favor one eye over the other.
The midface is the area between the eyes and the upper teeth. In children with Apert syndrome, this part of the face is smaller than usual and doesn’t grow in the typical way. The deepest point of the face is often the top of a shortened nose. Because the midface doesn’t grow forward normally, with growth being affected in all directions, many children will need surgery to bring this area forward, sometimes more than once as they grow. Measurements from our center suggest that the midface in children with Apert syndrome grows forward at only about one‑third the normal rate, and another research study from our Center found that forward growth may stop around age nine (Publication #25). A smaller midface can make the eyes look larger and can also contribute to breathing difficulties because the nasal passages are very narrow.
The Ears
In children with Apert syndrome, the ears are usually shaped normally, though they may sit a little lower on the head. Because the middle part of the face doesn’t grow as it typically would, the Eustachian tubes, which help to balance pressure on both sides of the eardrum, don’t work as well. This often leads to fluid building up behind the eardrum, making it hard to hear and causing more frequent ear infections. Most infants need small ear tubes to help with drainage and to prevent repeated infections or long‑term scarring of the eardrum. As children grow, this drainage problem usually improves.
In addition, some of the tiny bones inside the ear may be partially fused together, which can cause conductive hearing loss. Many adults with Apert syndrome have some degree of hearing loss, so keeping a close eye on ear health during infancy and childhood is very important.
The Mouth, Palate, and Airway
Another issue related to the small midface is that the palate, or roof of the mouth, often has a narrow, high arch. This high arch pushes up the floor of the nose, which makes it hard for children to breathe through their noses. Because the back of the nose is pushed up, parents may notice that their toddler’s nose seems to “run” almost all the time. This can get better as children grow, but the long‑term solution happens in the teenage years, when the midface is brought forward and the bite is corrected. This surgery also helps open the nasal passages and improves breathing.
Children with Apert syndrome may have a cleft palate. In most cases, the cleft affects only the back part of the palate, called the secondary palate, and rarely reaches the front. At our center, we’ve found that about 25% of children have a clearly visible cleft, and another 25% have what’s called a submucous cleft. With a submucous cleft, the surface of the palate looks normal, but the uvula is split, and the deeper muscles are not in the right position. A submucous cleft usually doesn’t need treatment unless it causes speech problems. The remaining 50% of children with Apert syndrome, or half, do not have any type of cleft palate.
In recent years, we have changed our approach to treating clefts in children with Apert syndrome. We’ve found that delaying the repair for several years (see Treatment) can offer important advantages, particularly with respect to breathing, and this timing is now part of our updated treatment plan.
Parents often notice that their child is a “noisy breather,” especially during sleep. This usually happens because the midface is smaller in children with Apert syndrome, which makes the nasal passages very narrow and pushed backwards. When babies can’t move air easily through their noses, they may work harder to breathe at night. This can lead to frequent awakenings and, in some cases, may affect how much oxygen reaches the brain during sleep. Sometimes the windpipe, or trachea, in children with Apert syndrome can be slightly narrowed, which adds to breathing difficulties. Normally, the trachea is held open by C‑shaped rings of cartilage, kind of like the wire coil around a dryer vent hose. Because they are C‑shaped, these rings can flex and widen when we cough or take a deep breath. In Apert syndrome, some children have O‑shaped rings instead. These rings can’t expand the same way, which makes it harder for the trachea to open wider when needed. Children may also have reactive airways, which is similar to asthma. In these cases, a pediatric pulmonologist (a lung specialist) may recommend medications commonly used for asthma to help with breathing.
We believe that the first and most important test for a newborn with Apert syndrome is not an X‑ray or CT scan, but an overnight sleep study. This test helps measure how well the child breathes during sleep and whether they are getting enough oxygen at night.
The Heart
Fortunately, most children with Apert syndrome do not have major heart problems. Fewer than 10% are found to have any type of heart issue. When heart concerns do occur, they are usually small openings in the heart walls, called ASDs (atrial septal defects) or VSDs (ventricular septal defects). Much more rarely, a child may have a condition called an overriding aorta or may experience episodes of a fast heart rate. Because these issues can occur, it’s important for children with Apert syndrome to be evaluated by a pediatric cardiologist, who can check for any heart‑related concerns and recommend treatment if needed.
The Stomach and Intestines
Most children with Apert syndrome experience reflux (gastro‑esophageal reflux, or GER). This often happens because they work harder to breathe, which increases pressure in the stomach. As a result, stomach contents can flow back up into the esophagus and sometimes even reach the lungs. In more severe cases, this can cause a baby to briefly stop breathing and turn blue.
There are several tests that can help diagnose reflux, including:
- A pH probe done during an overnight sleep study
- A barium swallow test
- An upper endoscopy
Because reflux is so common in these infants, we often recommend considering reflux medication early on.
A small number of children with Apert syndrome (less than 7%) may also have a condition called intestinal malrotation, where the intestines are not positioned normally. When there are feeding concerns, this is usually diagnosed with a test called a barium swallow and, if present, needs to be corrected by a pediatric surgeon.
The Hands and Feet
One of the most recognizable features of Apert syndrome is the fusion of the fingers and toes, called syndactylies. When all babies develop in the womb, their fingers and toes start out fused together. Normally, a signal tells the hands and feet to separate, creating individual fingers and toes. In Apert syndrome, that separation signal doesn’t happen the way it should, so the fingers and toes remain joined. Apert‑related hand differences are classified based on how many fingers are fused:
- Type I hand (mildest form): The index, middle, and ring fingers are fused together.
- Type II hand: The thumb is also connected to the index finger by a thin bridge of skin.
- Type III hand (“rosebud hand,” more severe): All the fingers are fused, and the thumb is joined to the index finger in a more complex way. In rare cases, the thumb may be completely hidden under the skin of the palm.
As a general rule, when the fingernails are fused together into one large nail plate, it usually means that the bones underneath are also fused. In children with Apert syndrome, the index, middle, ring, and small fingers are typically missing the middle joint (called the PIP joint). As the fingers grow, they can start to curve or bend, which may cause them to cross over one another. This is known as clinodactyly. The thumb can also curve, usually bending away from the index finger.
In most children with Apert syndrome, the toes have separate nails, which means the bones underneath are usually not fused. When bony fusions do happen, they typically involve only a couple of toes. The big toes are often shorter than average and may curve outward, similar to how the thumbs grow.
Although the feet may look very wide, measurements show that the width is actually close to normal, the real difference is that the feet are shorter than usual. Because of this mismatch (normal width but short length), finding shoes that fit well can be challenging. Many families end up buying shoes that are a size too long just to get enough width.
As children grow, three common bumps may develop on the feet:
- One along the inside edge, near the big toe
- One along the outside edge, just behind the small toe
- One on the bottom of the foot, lined up with the second toe
The treatment of these bumps, and the syndactylies is discussed in the Treatment section.
Below are examples of the three types of syndactylies, based on the degree of attachment of the thumb. It is possible to get five-fingered hands in almost every child with just two operations.
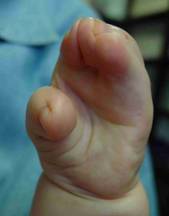
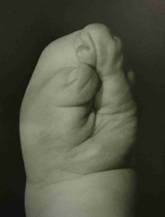

Type I Type II Type III
Other Joints
As children with Apert syndrome grow, parents may notice a few differences in how their joints move. Some children may not be able to lift their arms fully over their heads because the shoulder joints are tighter than usual. They may also have trouble fully straightening their elbows. The ankle joint is often positioned slightly more toward the inside of the foot rather than centered, which can affect how the foot moves. In addition, it’s common for some of the bones in the neck (the cervical vertebrae) to be fused together. At this time, there are limited surgical options to improve movement in these joints.
Treatment
The treatment of a child born with Apert syndrome is complex and is best provided by experienced craniofacial teams at major centers. Recent research from our Center suggests that we have the lowest published complication rate (2%) following operations to enlarge the head in children with syndromic craniosynostosis (Publication #98). This low complication rate compares very favorably, for example, with that of centers performing posterior skull distraction, which is associated with reported complication rates between 25% and 30%. The most common complication in our reviewed series was infection (6/498), which, interestingly, occurred only in secondary corrections. Our research also suggested that children who began treatment at other centers before transferring care to our Center had to undergo, on average, one additional skull surgery compared with those who began treatment in Dallas.
The following is a brief overview of how we care for our patients who come to Dallas for treatment. As a result of seeing a high number of children with Apert syndrome and conducting numerous research studies, our treatment approach differs significantly from what most other doctors might recommend. To begin with, our focus has shifted to maximizing intellectual development as the foremost priority. Our second goal is to significantly reduce the high number of operations that most children typically undergo.
Skull Surgery
The best time for the first skull operation depends largely on how the skull is growing. Our general rule of thumb is to delay skull surgery for as long as possible. Children treated in Dallas may have surgery delayed until 15 months of age, or even older, depending on multiple factors. The reasons for this delay are complex, but one concern is that our operations can impair skull growth, meaning that performing surgery too early may lead to additional procedures later. Our research (Publication #98) supports the premise that safely delaying surgery can eliminate the need for one additional skull operation. Therefore, we believe it is best not to operate before 11 months of age. As babies grow and get bigger, their total blood volume increases, reducing the likelihood that a blood transfusion will be necessary. The operation may also be less risky. We routinely recommend that children receive a drug called erythropoietin before surgery to raise blood levels. We have published a study showing that children who received this drug before surgery had a much lower risk of needing blood transfusions (Publication #20). We also use a “cell-saver” to recycle much of the blood lost during surgery so it can be returned to the child during the operation (Publication #23). Using this combination of techniques, most children, now well over 90%, do not require any blood transfusions. We believe that reducing blood transfusions also lowers the risk of other more serious complications.
The surgical team. At our center, operations on the skull are always performed by a pediatric neurosurgeon and a craniofacial surgeon working together throughout the entire procedure. We believe that having two experienced surgeons present at all times improves both the speed and safety of the operation. In addition, only pediatric anesthesiologists with extensive craniofacial surgical experience are involved. One of our pediatric ENT specialists will often evaluate the child before surgery, and if needed, place ear tubes at the same time as the skull surgery, thereby avoiding the need for an additional anesthetic.
Length of surgery. Typically, skull surgery takes two hours, but children are in the operating room for a total of 4 to 4½ hours. Whether the front half of the skull is brought forward (anterior CVR or FOA) or the back half is moved further backward (posterior CVR) depends on the findings of a preoperative MRI scan (CT scans cannot show the most important areas of the brain). We do not shave any hair, although many centers still prefer to do so. Many years ago, I changed the typical straight-line incision to a wavy, zigzag incision after realizing that when children get their hair wet, it will part along a straight-line scar, making it obvious (Publication #10). Designing the incision in a wavy pattern helps to better hide the scar, especially when the hair is wet.
The goal of the skull surgery is to increase its size to give the brain more room to grow. Once a suture is fused shut, it cannot be “released” so it can begin to grow again. This is because it is impossible to surgically recreate a functioning suture. For this reason, it is essential that the surgery achieve a significant enlargement, otherwise little is accomplished.
Fixation. Surgeons use various techniques to hold the skull bones in place after moving them into position. We have previously reported that, because of the way the skull grows, when other surgeons use metal plates and screws to put the skull back together, these plates may eventually end up on the inside of the skull, with the screws poking into the brain. (Publication #12). While unaware of any cases in which this has caused a serious problem, we have nevertheless chosen to use only dissolving stitches to put the skull back together (Publication #21). With this technique, nothing artificial is left behind as the child grows. Dissolving plates and screws can also end up inside the skulls of growing infants and can weaken the skull bones, making subsequent operations more technically challenging. Rarely, in a very small percentage of cases, the dissolving plates and screws may melt into a liquid that can drain out through a small hole in the overlying skin. Therefore, although it is technically a bit more difficult to rebuild a skull using only dissolving sutures, we believe this technique provides the best results with the fewest complications.
Many years ago, with the hope of being able to enlarge the skull more than with traditional skull surgery, I evaluated the use of distraction devices. These devices have metallic rods that protrude from the head, which parents must turn daily to slowly expand the skull. I found that skull distraction was not only difficult for both the child and the parents, but it also required two operations instead of one (a second operation is needed months later to remove the device). I also found that distraction deformed the skull, had a higher complication rate, and did not appear to provide any better result than a single operation (Publication #53). Therefore, I no longer recommend this technique for my patients. I also believe that helmets should never be used on a child with craniosynostosis, as these restrict skull growth, which is something that should never be done in Apert syndrome.
At the end of the operation in Dallas, the scalp is closed with dissolving stitches. We never use metal staples, nor do we use drainage tubes, because both are painful when removed. We also do not place any bandages on the child. Instead, we simply wash the hair and comb it out before leaving the operating room.
Prominent eyes. The most critically important step at the end of an operation to enlarge the front half of the skull (anterior CVR or FOA) for children with prominent eyes is for the surgeon to place a dissolving suture to keep the eyelids partly closed—but not so much that the child cannot see. Unfortunately, we have seen several children operated on at other centers who did not receive these eyelid stitches and, as a result of postoperative swelling, ended up completely losing vision in one or both eyes.
Hospital stay. Children typically spend one night in the pediatric intensive care unit before being transferred to the floor the following day. We encourage parents to hold their child in their laps rather than keeping them in a crib after surgery. Today, no narcotics are given to children after surgery; instead, we keep them comfortable using intravenous acetaminophen and ibuprofen (Publication #58), which we have also found reduces nausea and vomiting. At our center, nearly all children spend only two nights in the hospital before discharge. The risks of surgery are very small at experienced centers. Many studies have shown that surgeons with the most experience tend to have the fewest complications (Publication #37). We have published a two‑center study (Publication #15) showing that no infections occurred in infants undergoing operations for the first time (although it is still possible for this to occur).
The average child with Apert syndrome usually needs more than one skull operation as they grow. It is very important that children be followed closely into their late teenage years to monitor for raised intracranial pressure, optic nerve atrophy, and the development of a Chiari malformation, which can progress with age.
Breathing and Sleep Apnea
The most significant factor affecting mental development in Apert syndrome is not elevated intracranial pressure, but rather the amount of oxygen the brain receives during sleep. Difficulty breathing while asleep is known as sleep apnea, which occurs in two forms: central and obstructive.
- Central apnea is when the brain “forgets” to breathe. This is often caused by cerebellar tonsillar herniation (a Chiari), or less frequently from raised intracranial pressure. One treatment for central apnea is to enlarge the back of the skull, while simultaneously enlarging the bone around the upper spine to decompress the Chiari (Publication #48).
- However, obstructive apnea is the most common type of apnea in Apert syndrome. It is typically the result of a narrowed or blocked airway (most often, compressed nasal passages). If a child has mild obstructive sleep apnea, medication might be recommended as a first step. For more moderate apnea, a tonsillectomy might be considered, depending upon their size. The last option, short of major surgery, is to try CPAP (Continuous Positive Airway Pressure). This entails being fitted for a face mask, which is worn at night. Each time the child inhales, a gentle pressure is applied to help air reach the lungs. CPAP is typically very effective when used throughout the night; however, many parents find that their child removes the mask at some point, which can reduce the treatment’s overall success. Fortunately, there are some tricks to help children adjust.
If none of the above treatments are able to successfully provide healthy oxygen levels at night, then surgery might be necessary. Depending on a child’s age, either a tracheostomy or a midfacial advancement might be recommended. In Dallas, we believe that temporary tracheostomies are the safest option with the best overall outcomes for children under 7 years of age. For older children, however, we will consider a midfacial advancement (see below).
Surgery of the Midface
The midface describes the area between the upper teeth and the top of the nose. There are three basic operations used to bring the midface forward in Apert syndrome: the LeFort I, the LeFort III and the monobloc.
- The LeFort I brings the lower midface forward from the level of the upper teeth to just below the nostrils. However, this operation is usually reserved until children have completed their growth in teenage years.
- The LeFort III brings the entire midface forward in one piece from the upper teeth to the top of the nose, including the cheekbones.
- The monobloc brings the forehead and the midface forward at the same time.
Which operation is best? Some surgeons will recommend a monobloc procedure for their patients with Apert syndrome. In our view, while advancing both the forehead and midface at the same time may theoretically spare a child an additional operation, there are several reasons we do not offer this procedure to our patients. First, the optimal timing for advancing the forehead does not align with the optimal timing for advancing the midface, yet the monobloc is doing both at the same time. More importantly, research has shown that the monobloc carries a high risk of serious infection (Publications: #8, #66). This may be why the LeFort III is currently the most commonly performed operation for addressing midfacial issues in Apert syndrome. Although it is a major procedure, the LeFort III has the greatest impact on normalizing a child’s appearance and improving nighttime breathing. In our view, however, many surgeons perform this operation too early; some studies indicate the average age elsewhere is around 5 years. Our research shows that midfacial growth stops after this procedure, meaning that when it is done at a young age, a second operation is often required later in childhood (Publication #55). Based on our findings, delaying surgery until after age 8, and applying an appropriate degree of overcorrection, makes it very unlikely that a child will ever need a repeat LeFort III.
The LeFort III is performed using the same incision on the top of the child’s head that is used for enlarging the skull. During this procedure, the bones of the midface are released across the top of the nose, then along the floor of the orbits (under the eyes), and finally down each side of the cheekbones. No scars are put on the child’s face. With the traditional LeFort III, after the bones were loosened the midface would be moved forward as a unit and held in place with bone grafts (taken from the skull) and metallic plates and screws. In younger children, the teeth were wired together for 4-6 weeks. However, with our updated technique, bone grafts are unnecessary, and we do not use any metal plates or wire the teeth together.
Dr. Fearon first developed the halo-distraction technique for the LeFort III back in 1998. This technique utilizes a device called the RED (Publications #18, #25, #55), which is actually not red, but is purple in color. It gets its name from being a Rigid External Distraction device. The RED device is used primarily in growing children. With the RED procedure, the bones of the midface are loosened and then instead of pulling the midface forward on the table, and then filling in the gaps with skull bone, the scalp is instead closed, and a halo is attached to the outside of the head with 8-10 screws. Next, two wires are attached, under the upper lip (so there are no scars on the face), to the facial bones, which then extend to the halo. By turning screws daily on the halo, the midface is slowly (and painlessly!) brought forward. The children are allowed to eat soft foods, may go to school, and can even go swimming while wearing the RED. For some children wearing this device after surgery is easier than for others. Although some surgeons suggest their patients wear the RED for up to 3 months, at our Center they typical length of time is just 7 to 8 weeks. The RED is then removed under a brief anesthetic.
One advantage of using the RED device is that it requires a smaller operation than a traditional LeFort III. However, the greatest benefit is its ability to move the midface significantly farther forward than is possible with the traditional technique. We have treated more than 200 children with this approach, and our extensive experience has allowed us to continually refine and improve the surgical technique.



Two different children, picture above, with Apert syndrome undergoing a halo-distraction LeFort III. Other pictures are available in our published studies.
Treating the Hands and Feet
Usually, the first thing new parents of a child with Apert syndrome notice is that they cannot count individual fingers and toes. This is because they appear fused together, a condition known as syndactyly. In typical fetal development, all fingers and toes begin fused, and later a chemical signal triggers their separation. In Apert syndrome, this separation signal does not occur. While children without Apert syndrome may occasionally be born with one or two fused fingers or toes, in Apert syndrome the fusion affects all fingers and toes on both hands and feet.
The surgery required to separate fused digits (syndactyly releases) is more complex in children with Apert syndrome than in those with isolated syndactyly. At our center in Dallas, we separate all ten fingers and toes in just two operations. Unlike some other centers, we believe that no child should need more than two procedures to achieve full separation, and that no fingers should ever be discarded; a practice that, unfortunately, still occurs. Our experience has shown that having a five‑fingered hand becomes increasingly important as children grow older (Publication #22). If a surgeon tells you that achieving a five‑fingered hand is not possible, it may be worthwhile to seek a second opinion from a center with more extensive experience. Apert syndrome is uncommon and most centers have very little experience treating this condition.
We have also observed that many surgeons are either unable or unwilling to separate toes. While toe separation does not significantly improve a child’s ability to walk, and some may argue that the only functional benefit is the ability to wear flip‑flops, we have heard from many parents that the impact goes far beyond this. Children who do not undergo toe separation often become self‑conscious as they grow older, avoiding swimming pools, beaches, or even taking off their shoes to run in the grass because they feel embarrassed by the appearance of their fused toes. In reality, separating the toes provides a meaningful functional benefit by allowing children to participate more comfortably in everyday activities and feel more at ease socially.
A common misconception is that separating the toes will widen the foot. Our research shows that the skin bridges between the toes do not keep the toes closer together and releasing them does not increase foot width. Toe width is determined entirely by the underlying bone structure. We have also found that the toes do not grow forward normally, and this limited forward growth alters the foot’s length‑to‑width ratio, making the foot appear wider than it actually is.
In Dallas, finger and toe separations are performed by a highly experienced team, allowing us to complete the release of all ten fingers and toes in just two operations. Many other centers recommend three or more procedures for the fingers alone; this not only exposes the child to unnecessary surgeries, but also increases the number of anesthetics, which may have negative effects on the developing brain. For these reasons, we limit the releases to two stages. Children spend only one night in the hospital and return home the following day. They are placed in casts that can be unwound at home two weeks later. Changing bandages after the casts come off can help optimize the final outcome.
Three months after the first-stage release, the second-stage procedure is performed to complete the separation of all fingers and toes. For most children, skull surgery is scheduled approximately three months after the second-stage syndactyly release. However, this sequence may vary depending on the child’s specific skull type.
Once children reach about 10 years of age, additional surgery can be performed to improve both the function and appearance of their hands. During this procedure, bends are created in the middle of each finger to mimic the appearance (but not the motion) of a PIP joint, and the thumbs are straightened. These changes enhance fine motor skills and help the hands look more natural. This surgery often enables children to button their shirts, use zippers, and tie their shoes more easily. At the same time, any painful bumps on the feet can be reduced, and angled toes can also be straightened.
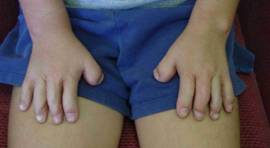
Examples (below) of released hands (top and middle row, Type I hands; bottom row, a released Type III hand with bends put in the left hand already).




Examples (below) of a patient with un-released toes, and one in whom the toes that have been separated:


Another reasons to release the toes (below).



The Big Picture
Most children with Apert syndrome undergo far more operations than necessary and spend too much time in the hospital. With recent studies raising concerns about the potential effects of multiple anesthetics on the developing brain, it is even more important to limit the number of procedures. Too often, children with Apert syndrome are taken to the operating room for a single small procedure performed by one specialist who is not coordinating with others, resulting in multiple surgeries each year.
Whenever a child with Apert syndrome requires anesthesia, it is essential that all specialists work together to accomplish as much as possible during the same session. Ideally, families should have a single “quarterback” who oversees and coordinates care among all the different subspecialists to ensure the most efficient and thoughtful treatment plan.
Our experience treating children with Apert syndrome has shown us that the primary goal of care is to prevent avoidable developmental delays, giving each child the best possible chance to lead a normal life. It is equally important that the correct operation be performed the first time and that every effort is made to minimize complications. Operating too early may seem beneficial, but it often disrupts normal growth and leads to additional procedures later on.
It is not uncommon for us to meet children who began treatment at other centers yet have made little progress. They have endured the discomfort of surgery, only to require the same procedure again because it was performed too early or not carried out in the most effective way. We believe strongly that children should be given substantial periods away from the hospital so they can grow, develop, and thrive to their fullest potential.
Ideally, parents should bring their child to the most experienced craniofacial centers they can access. In general, the busier the surgeon, the fewer the complications and the better the outcomes. Treatment recommendations evolve over time and can differ from one center to another, so it is important to discuss the overall treatment plan thoroughly with your doctor and ensure that all your questions are fully answered (see Choosing a Doctor).
Take the time to learn as much as possible, seek more than one opinion, and continue searching until you find a physician who has both the experience to achieve the safest, best results and the commitment to coordinate all the specialized care your child will need.
Jeffrey A. Fearon, MD
Director, The Craniofacial Center, Dallas Texas
972-566-6464
cranio700@thecraniofacialcenter.com